





























Upon injury or illness, the human body has an impressive repertoire of machinery in place for self-healing. One important protective mechanism for tissue regeneration and wound healing is the formation of extracellular matrix (ECM). Fibrosis is a dangerous condition where an excessive accumulation of ECM, in response to chronic injury or illness, leads to organ disfunction and failure (Jun et al., 2018). The United States government estimates that 45% of deaths in the US can be attributed to fibrotic disorders (Wynn et al., 2004) and some examples of potentially fatal fibrotic diseases include idiopathic pulmonary fibrosis, renal fibrosis (chronic kidney disease), hepatic cirrhosis, and cardiac fibrosis. Currently, there are few treatments that can halt disease progression.
1. Fibrosis relies on activation of wound healing fibroblast cells
Fibroblasts are specialized wound healing cells found in all organs and are responsible for regulating the ECM. Fibroblasts are activated by cytokines expressed at the injury site, such as TGF-β1, which result in their differentiation into myofibroblast cells. The myofibroblasts produce a large amount of ECM protein that give the injury site structural integrity during repair and regeneration (Jun et al., 2018). The ECM, composed of proteins such as collagen and fibronectin, is then degraded by metalloproteases (MMPs) and remodeled to rebuild the parenchymal tissue architecture. However, repeated injury or chronic inflammation does not provide enough time for the ECM to be resolved and the accumulation of ECM can form fibrotic lesions (Jun et al., 2018).
Example of fibroblasts involvement in fibrosis pathogenesis
2. Therapeutic strategies for reversing fibrosis
There is growing evidence that fibrosis may be reversible with the right treatments. Currently, there are three approaches to targeting and reversing fibrosis. The first is to eradicate the injury or source of inflammation; which is possible for fibrosis caused by viral infections (such as HCV and HBV for liver fibrosis) through antiviral therapy. However, eradication of the injury is not always feasible (Jun et al., 2018).
The second method for reversing fibrosis is to directly target and degrade the fibrotic ECM. This can be achieved by increasing MMP activity. TIMPs (tissue inhibitors of metalloproteinases) are a potential target to regulate MMPs as their increased expression level has been shown to correlate with the degree of cardiac and liver fibrosis. (Vanhoutte et al., Jun el al). Another potential target is transglutaminase 2 (TG2), which can cross-link ECM components collagen and fibronectin, making them more resistant to breakdown (Olsen et al., 2011).
Finally, eliminating or inhibiting the function of the pro-fibrotic myofibroblast cells may be used as an effective therapeutic strategy. There are many studies trying to better understand the signaling pathways and gene expression of proteins important for the function and survival of myofibroblasts (Jun et al., 2018).
3. Growing evidence for the use of relaxin for fibrotic diseases
The peptide hormone relaxin is known for reducing organ fibrosis in a variety of experimental models (Kanai et al., 2019). Relaxin acts through its cognate G protein coupled receptor (GPCR), RXFP1, to inhibit ECM production and can even reduce established scarring by degrading aberrant ECM through increased MMP expression (Kanai et al., 2019). The effects of relaxin (and the recombinant form serelaxin) are mediated via the nitric oxide pathway, which promotes the formation of cGMP and inhibits TGF-β1 profibrotic signaling pathways (Lv et al., 2018). (Figure 2). Serelaxin has shown promise in a variety of clinical trials; however, so far, none have been ultimately successful. This may be due to organ- and disease-specific issues concerning the delivery and pharmacokinetics of serelaxin (Kanai et al., 2019). Over the past few years, there has been a growing interest in better understanding the mechanism of action of relaxin in hopes of using it as a successful therapeutic.
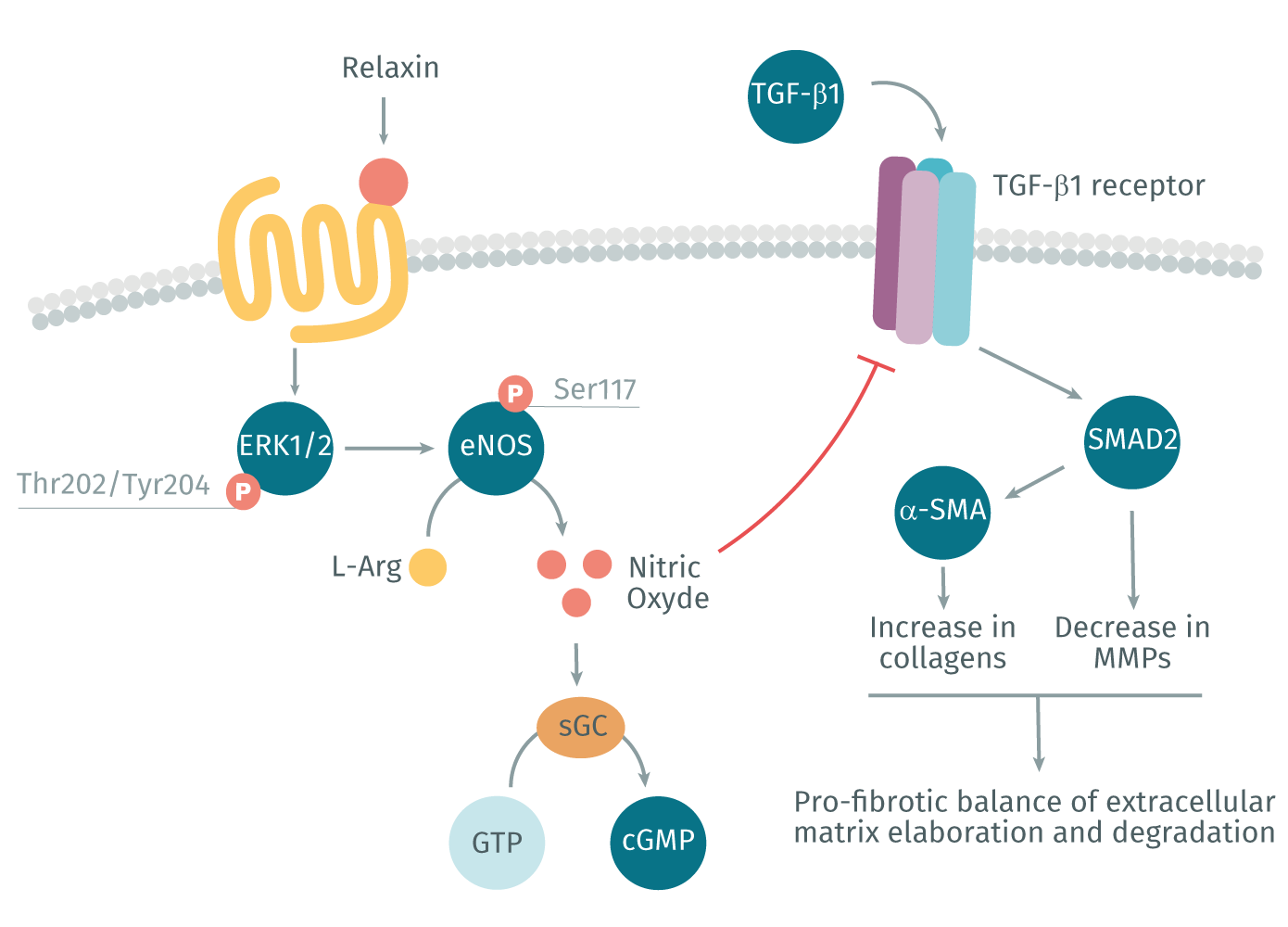
Relaxin signaling via RXFP1 and the nitric oxide pathway promotes the inhibition of TGF-β1 pro-fibrotic signaling pathway
Recently, Chow et al. (2019) used both the AlphaScreen™ SureFire pERK detection kit and HTRF™ cGMP kit to investigate signaling pathways activated in response to recombinant relaxin treatment of rat renal myofibroblasts and human cardiac myofibroblasts. Previous studies had suggested that the effect of relaxin required the presence of angiotensin receptors. Therefore, Chow et al. sought to understand the involvement of the AT1R and AT2R angiotensin receptors in the anti-fibrotic response of relaxin. They found that the antifibrotic effects induced by relaxin could also be inhibited by an AT1R antagonist, even though relaxin does not bind the receptor directly. This suggests a crosstalk between the three GPCRs (AT1R, AT2R, and RXFP1) playing a relevant role in the balance between pro- and anti-fibrotic pathways. These findings have significant therapeutic implications as targeting this receptor crosstalk may be the key to the discovering an effective treatment for fibrosis.
Read more on how HTRF, and AlphaLISA and other Revvity technologies have been used to advance fibrosis related research by helping researchers better understand the underlying mechanisms of the disease.
For research use only. Not for use in diagnostic procedures.
- Chow BSM, Kocan M, Shen M, et al. AT1R-AT2R-RXFP1 Functional Crosstalk in Myofibroblasts: Impact on the Therapeutic Targeting of Renal and Cardiac Fibrosis. J Am Soc Nephrol. 2019;30(11):2191-2207. doi:10.1681/ASN.2019060597
- Jun JI, Lau LF. Resolution of organ fibrosis. J Clin Invest. 2018;128(1):97-107. doi:10.1172/JCI93563
- Kanai AJ, Konieczko EM, Bennett RG, Samuel CS, Royce SG. Relaxin and fibrosis: Emerging targets, challenges, and future directions. Mol Cell Endocrinol. 2019;487:66-74. doi:10.1016/j.mce.2019.02.005
- Lv W, Booz GW, Fan F, Wang Y, Roman RJ. Oxidative Stress and Renal Fibrosis: Recent Insights for the Development of Novel Therapeutic Strategies. Front Physiol. 2018;9:105. Published 2018 Feb 16. doi:10.3389/fphys.2018.00105
- Olsen KC, Sapinoro RE, Kottmann RM, et al. Transglutaminase 2 and its role in pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184(6):699-707. doi:10.1164/rccm.201101-0013OC
- Vanhoutte D, Heymans S. TIMPs and cardiac remodeling: ‘Embracing the MMP-independent-side of the family’. J Mol Cell Cardiol. 2010;48(3):445-453. doi:10.1016/j.yjmcc.2009.09.013
- Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol. 2004;4(8):583-594. doi:10.1038/nri1412


