


































Metagenomics, a term coined to describe the comprehensive analysis of microbial genomes in environmental samples, has changed microbial ecology. Traditionally, microorganisms have been studied through the culturing of individual species or strains using artificial culture media; however, it has been estimated that less than 2% of bacteria can be cultured in the laboratory. As a result, our understanding of the genetic diversity of microorganisms in the environment is just scratching the surface.
The technologies and methods used in metagenomics allow direct analysis of the genetic material of the community present in the sample, without the need for individual culture and isolation. It has multiple applications, ranging from identification of pathogenic microorganisms in infectious diseases, monitoring resistance and virulence, studies of microbial diversity and dynamics to surveillance of food production workflows.
In this blog we will provide a step-by-step guide of the different techniques and challenges that are common in this area.
Sample collection Regardless of the sample type that we are considering, the most representative snapshot of the community we want to study is obtained by collecting a sample and either immediately processing it or snap-freezing it in liquid nitrogen and transferring it to -80°C for long-term storage. This might not always be possible, especially when collection takes place outside the lab or in remote locations. Alternatively, the sample can be placed in a preservation buffer to make it more stable at room temperature and preserve nucleic acid integrity.
Sample homogenization Gram-negative and Gram-positive bacteria have very different cell wall structures, so the challenge is to obtain a complete lysis of all microbes present in the sample. Incomplete lysis can lead to misrepresentation of the microbial community we want to study. For metagenomics, an efficient and popular method is mechanical lysis using bead beating. There are a few other methods available, including chemical lysis (e.g. lysozyme), sonication and thermal lysis. Some researchers advocate for a combination of mechanical and chemical procedures but ultimately the method of choice will depend on the sample type we are working with and the bacterial mass that we estimate to find (e.g. high mass in stool vs low mass in skin swabs).
Nucleic acid extraction: Isolation of DNA or RNA is a crucial step because the nucleic acids are the input material for any study downstream. There is a strong correlation between the quality of the DNA and RNA used and the quality of the data obtained. There are several features to consider when choosing a suitable method:
- Yield: The amount of material available for extraction can be very variable, so its desirable to use an efficient method for extraction, which will increase chances of obtaining enough yield for downstream library preparation. It is important to bear in mind that an inefficient extraction method can lead to additional biases in the types of microorganisms that are detected.
- Purity: The presence of impurities and PCR inhibitors (e.g. residual proteins, polysaccharides, phenol, chloroform and salts) is common in metagenomics samples, particularly in stool and soil. If not removed by the extraction method they can interfere with enzymatic reactions downstream, leading to poor sequencing results. Typically, the best purity results require specialized chemistry or methodology to produce high quality results.
- Integrity: Damaged (single strand breaks) or fragmented nucleic acids can lead to missasembly of genomic sequences, reduced detection of microorganisms and errors in taxonomic in functional annotation. In long-read sequencing, the demand for high concentrations of highly purified DNA of high molecular weight (HMW) is even more pronounced.
Library preparation Following DNA or RNA extraction, library preparation is performed. The specific workflow to be used will depend on the specific question we want to answer, but we can distinguish three groups:
- 16S rRNA sequencing used when we only want to identify and quantify the bacteria present in each sample. The 16S gene contains nine variable regions, designated as V1 through V9, interspersed between conserved regions. The variable regions provide the basis for differentiation between genus or species. The most popular regions are V4, V3-V4 and V1-V3, depending on specific research goals, sample types, what has been done in previous studies, etc. Compared to whole genome sequencing, 16S rRNA sequencing generally requires less input DNA and is less impacted by the presence of host DNA. It is also more cost-effective (100,000 reads per sample is sufficient for most metagenomic surveys) and robust.
- Whole genome sequencing used when we want to have a complete picture of all the genomes present in the sample. For example, it can be the method of choice when looking for antibiotic-resistance genes, or a specific metabolic pathway. The workflows existing in the market are divided in those that use mechanical fragmentation and those that use enzymatic fragmentation of the genomic DNA. In the past methods using mechanical fragmentation where preferred because it was assumed to be more random, but current fragmentases present very little bias and are more popular now, due to the convenience of this method.
- Metatranscriptomics or gene expression analysis of the whole community. The major challenge here is the presence of rRNA, that will hamper detection of low-abundance transcript. Removal of rRNA was challenging, but currently exists approaches, such as the use of CRISPR-Cas9 system, that can efficiently deplete these uninformative molecules from both host and all the species of the community in a single step.
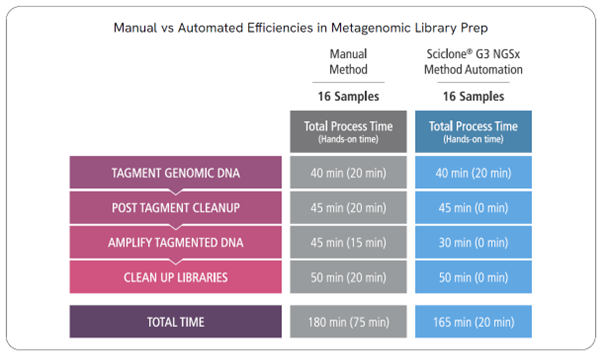
Automation of an NGS-based metagenomics library preparation offers significant advantages over manual sample preparation. Increased throughput and scalability, reduction in human touchpoints and human error, enhanced consistency and reproducibility, and increased speed all contribute to reliable data production that is amenable to varying throughputs for metagenomics labs.
Library prep automation
- Sequencing Illumina next-generation sequencing (short read) is still the technology most frequently used for metagenomics studies. However, over the last years new sequencing methods using long-read (e.g. Oxford Nanopore Technologies, PacBio) have become more frequent.
- Data analysis: After sequencing, raw reads are processed through bioinformatics pipeline. Many of the limitations associated to the lack of robust data analysis tools are discussed in this blog post.
For research use only. Not for use in diagnostic procedures.
References:
- Wade, W (2002) Unculturable bacteria – the uncharacterized organisms that cause oral infections. J. R. Soc. Med. 95(2): 81–83.
- Hoopen, P. et al (2017). The metagenomic data life-cycle: standards and best practices, GigaScience, 6 (8).
- Zhang, D., Lou, X., Yan, H. et al. (2018) Metagenomic analysis of viral nucleic acid extraction methods in respiratory clinical samples. BMC Genomics 19, 773.
- Zhenli Diao, Yuanfeng Zhang, Yuqing Chen, Yanxi Han, Lu Chang, Yu Ma, Lei Feng, Tao Huang, Rui Zhang, Jinming Li (2023). Assessing the Quality of Metagenomic Next-Generation Sequencing for Pathogen Detection in Lower Respiratory Infections, Clinical Chemistry, 69 (9) 1038–104.
- Steve M., Charles C. (2022). The Role of Metagenomics and Next-Generation Sequencing in Infectious Disease Diagnosis, Clinical Chemistry, 68(1) 115–124.
- Zhang B, Brock M, Arana C, Dende C, van Oers NS, Hooper LV, Raj P (2021). Impact of Bead-Beating Intensity on the Genus- and Species-Level Characterization of the Gut Microbiome. Front. Cell. Infect. Microbiol 11
- Trigodet, F., Lolans, K., Fogarty, E., Shaiber, A., Morrison, H. G., Barreiro, L., Jabri, B., & Eren, A. M. (2022). High molecular weight DNA extraction strategies for long-read sequencing of complex metagenomes. Molecular Ecology Resources, 22, 1786-1802
- Improving the efficiency of metagenomic analysis of stool samples. (Revvity Technical Note, AG011805_24_TN)


